
El síndrome de VEXAS es una enfermedad poco frecuente y de reciente descripción. Combina manifestaciones inflamatorias y hematológicas, lo que suele complicar su diagnóstico inicial. En Mapfre queremos ofrecerte una visión clara de sus características principales para facilitar su reconocimiento temprano.
¿Qué significa VEXAS?
El nombre de esta patología es en realidad un acrónimo que define sus bases biológicas:
- V (Vacuolas): Presencia de cavidades en las células precursoras de la sangre.
- E (Enzima E1): Afectación de la enzima necesaria para el metabolismo de proteínas.
- X (Ligado al cromosoma X): El gen implicado se encuentra en este cromosoma.
- A (Autoinflamatorio): El cuerpo genera una respuesta inflamatoria sin causa externa.
- S (Somático): La mutación no se hereda, se adquiere durante la vida.
Origen de la enfermedad
A diferencia de otras condiciones genéticas, el síndrome de VEXAS no es una enfermedad hereditaria. Su origen reside en una mutación somática espontánea en el gen UBA1, que ocurre en las células madre de la médula ósea durante la edad adulta. Aunque es una patología identificada muy recientemente (año 2000), su estudio ha avanzado exponencialmente.
¿A quién afecta?
El perfil principal del paciente son varones mayores de 50 años.
- Edad media de inicio: 64 años (rango habitual entre 40 y 85 años).
- Excepciones: Se han documentado casos aislados en mujeres y personas menores de 40 años.
Dada su reciente descripción, es muy común que estos pacientes hayan sido tratados previamente por otras dolencias antes de recibir el diagnóstico correcto. Es fundamental contar con un buen seguro de salud que permita el acceso a especialistas para un seguimiento exhaustivo.
Manifestaciones clínicas
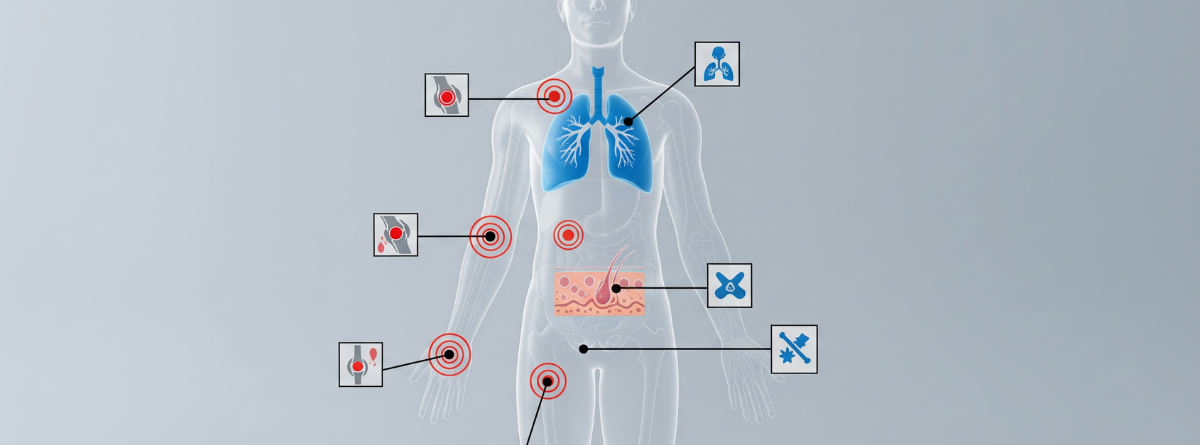
Esta enfermedad provoca una inflamación sistémica que puede afectar a casi cualquier órgano. Las manifestaciones más comunes se dividen en tres áreas:
| Tipo | Síntomas comunes |
| Inflamatorías | Fiebre recurrente, dolor articular, condritis (inflamación del cartílago en nariz o orejas). |
| Dermatológicas | Pápulas y nódulos de color eritemato-violáceo en la piel. |
| Hematológicas | Anemia macrocítica, plaquetas bajas (trombocitopenia) y riesgo de síndromes mielodisplásicos. |
Además, los pacientes suelen experimentar una fatiga intensa, pérdida de peso e infiltrados pulmonares.
Diagnóstico
El diagnóstico es un reto médico y se apoya en tres pilares fundamentales:
- Sospecha clínica: Varones con inflamación persistente, marcadores elevados (PCR, ferritina) y anemia macrocítica sin infección aparente.
- Manifestaciones específicas: Como la condritis auricular o lesiones cutáneas dolorosas.
- Dependencia de corticoides: Pacientes que recaen inmediatamente al bajar la dosis de medicación.
La confirmación definitiva se realiza mediante una prueba genética para identificar la mutación en el gen UBA1. Ante síntomas persistentes y complejos, disponer de una segunda opinión médica puede ser clave para orientar el caso.
Tratamiento
Actualmente no existe una cura definitiva, por lo que el objetivo médico es controlar la inflamación y prevenir daños mayores.
- Corticosteroides: Son la primera línea de choque, aunque su uso prolongado puede causar diabetes o pérdida de densidad ósea.
- Terapias biológicas e inmunosupresores: Como los inhibidores de JAK (ruxolitinib), que reducen la dependencia de los corticoides.
- Agentes hipometilantes: Utilizados si aparece un síndrome mielodisplásico asociado.
- Trasplante de médula: En casos seleccionados, se considera el trasplante de progenitores hematopoyéticos.
Nuevas investigaciones (2024-2025) exploran moléculas como la TAK-243, que busca eliminar selectivamente las células mutadas.
Pronóstico
Al ser una enfermedad «joven» para la ciencia, el pronóstico sigue en estudio. Se considera una patología progresiva que requiere un manejo multidisciplinar constante para minimizar las recaídas y los efectos secundarios de la medicación.
Contar con el respaldo adecuado es fundamental cuando nos enfrentamos a diagnósticos complejos. En Mapfre te ofrecemos seguros de salud con acceso a los mejores especialistas y las pruebas diagnósticas más avanzadas, para que tú y tu familia estéis siempre en las mejores manos.
Importancia del conocimiento
La detección precoz del síndrome de VEXAS es vital para mejorar la calidad de vida del paciente. Divulgar su existencia ayuda a que más profesionales y pacientes identifiquen los síntomas antes de que la enfermedad avance.
LO QUE DEBES SABER…
- El síndrome de VEXAS es una enfermedad autoinflamatoria rara causada por una mutación somática en el gen UBA1, adquirida en la edad adulta y no hereditaria.
- Se caracteriza por una combinación de manifestaciones inflamatorias, cutáneas y hematológicas, con gran variabilidad clínica y frecuente dificultad diagnóstica.
- No existe un tratamiento curativo, por lo que el manejo se centra en controlar la inflamación y las complicaciones, siendo una enfermedad progresiva con pronóstico aún incierto.
Contar con el respaldo adecuado es fundamental cuando nos enfrentamos a diagnósticos complejos. En Mapfre te ofrecemos seguros de salud con acceso a los mejores especialistas y las pruebas diagnósticas más avanzadas, para que tú y tu familia estéis siempre en las mejores manos.






