Síndrome de Rokytansky, ¿qué es?

Resumen del contenido

Dra. Natalia García Montaner
Médico especializada en ginecología y obstetricia. Con más de dos décadas de experiencia en la profesión, es una experta en ecografía de 20 semanas, diagnóstico prenatal, parto, histeroscopia quirúrgica y laparoscopia, entre otras. Compagina su trabajo en diferentes centros con la docencia y la redacción de artículos.
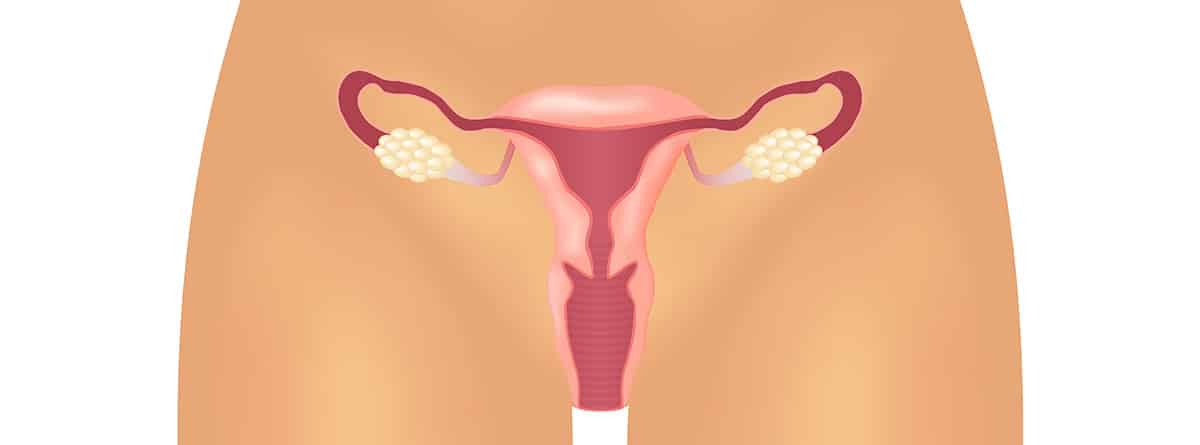
El síndrome de Mayer-Rokitansky-Küster-Hauser (MRKH), conocido habitualmente como síndrome de Rokitansky, es una anomalía congénita del aparato reproductor femenino. Debe su nombre a los cuatro médicos que hicieron su descripción y clasificación. Mayer, ginecólogo y anatomista alemán, y Rokitansty, patólogo austriaco, publicaron a principios del siglo XIX las primeras descripciones de mujeres con este síndrome, los doctores Küster y Hauser, en la segunda mitad del siglo XX aportaron nuevos datos y publicaron una descripción completa de esta patología.
Se trata de una malformación congénita que afecta a una de cada 4.500 niñas en la que embriológicamente no se ha desarrollado el útero y los dos tercios superiores de la vagina. El resto de aparato genital interno y externo es completamente normal: vulva de aspecto normal y ovarios normales y funcionales que realizan perfectamente su función, tanto la de producir óvulos como su función endocrina de secreción hormonal con lo que llegada la adolescencia el desarrollo de las características sexuales secundarias: cambios en el aspecto de la vulva, mamas, vello corporal, distribución de grasa corporal… se producen con total normalidad, el hecho diferencial es que la regla nunca aparece pues el órgano que sangra durante la regla no existe. Esto hace que el diagnóstico se produzca siempre durante la adolescencia pues son niñas cromosómicamente normales, con fórmula cromosómica 46XX, con genitales externos completamente normales tanto en la infancia como en la edad adulta, pero que al retrasarse la edad de la primera menstruación consultan al ginecólogo y las pruebas de imagen (ecografía y/o resonancia magnética) confirman que hay una ausencia congénita del útero. Incluso el tercio externo de la vagina es completamente normal, aunque a una distancia muy corta acaba en un fondo de saco, por lo cual, sin tratamiento no pueden mantener relaciones sexuales con penetración vaginal.
¿Qué causa tiene?
La causa es desconocida; hay un factor genético implicado pues se dan casos en la misma familia y se ha descrito alguna mutación en genes concretos responsables de que en el embrión femenino entre la semana 4 y 12 de gestación se produzca un error en el desarrollo del útero y la parte superior de la vagina que en algunos casos se asocia a otros órganos. A este respecto hay descritos dos tipos de síndrome de MRKH:
- Tipo I, en el que de forma aislada la anomalía afecta únicamente al aparato reproductor
- Tipo II, en el que se asocia a otras malformaciones:
- Riñones en el 40% de los casos.
- Columna vertebral en el 20-25 %.
- Oído en el 10%.
- Corazón en menor frecuencia.
¿En qué consiste el tratamiento?
- Apoyo psicológico cuando se requiera.
- Reconstrucción vaginal para que se puedan mantener relaciones sexuales con normalidad. Esto se puede conseguir de dos modos, con unos dilatadores que, usándolos a diario, con el tiempo consiguen alargar la longitud de la vagina, y cuando esto no funciona se puede reconstruir una neo-vagina quirúrgicamente.
¿Qué consecuencias tiene?
Las opciones de maternidad se reducen a tratamientos de maternidad subrogada con óvulos propios que son perfectamente normales, pero que tendrán que ser fertilizados in vitro y transferidos a un “vientre de alquiler”.
En 2014, en Suecia se dio el primer caso de una mujer con este síndrome en dar a luz tras un trasplante de útero. Este hito médico abrió una nueva esperanza a mujeres con esta condición, y desde entonces se han realizado más de 100 trasplantes de útero en todo el mundo. En España el primer caso es el de una mujer con síndrome de MRKH que fue sometida, en el Hospital Clínic de Barcelona, a un trasplante del útero de su hermana en 2020 y dio a luz por cesárea en 2023 a un bebé prematuro de 1.125 gramos que se desarrolló con normalidad.
¿Cómo actuar?
En cuanto se confirma el diagnóstico se debería hacer un estudio completo que incluya ecografía renal, radiografía de columna, ecografía cardiaca, audiograma, para poder diagnosticar, y tratar si es necesario, las anomalías asociadas en casos de síndrome MRKH tipo II
También hay que ofrecer consejo genético para valorar exactamente los riesgos de transmisión de esta anomalía a la descendencia y al resto de miembros de la familia.
Por lo demás, se trata de mujeres con cromosomas normales, desarrollo y aspecto completamente normal que pueden mantener una vida completamente normal sin alterar su calidad ni esperanza de vida.
En los Seguros de Salud de MAPFRE cuentan con grandes especialistas que te ayudarán al diagnóstico, tratamiento y seguimiento de la enfermedad.
Lo que debes saber…
- El síndrome de Rokitansky es una malformación congénita en la que falta el útero y parte de la vagina, pero los ovarios y características sexuales son normales.
- Tiene un componente genético y puede asociarse a malformaciones en otros órganos como riñones o columna.
- El tratamiento incluye apoyo psicológico y reconstrucción vaginal; la maternidad se logra vía subrogación o trasplante de útero.



Comentarios (0)